Abstract
Huntington Disease-like (HDL) is a neurodegenerative disorder similar to Huntington Disease (HD) in its clinical phenotype, genetic characteristics, neuropathology and longitudinal progression. We review the different phenocopies of HDL in Africa from a clinical and genetic perspective through published cases. A literature review through PubMed and Google Scholar of all clinically and genically described cases of HDL until the end of December 2022 was performed and a descriptive analysis was carried out. Fifteen papers were published from 2000 to 2022 in Africa on HDL. Only HD phenocopies caused by mutation of the following genes (JPH3, ATXN2, VPS13A, VPS13D, PRNP, NBIA, ATN1, ATM) were described. The most representative phenocopies was HDL2 (JPH3) described in case series and families in South Africa. Other phenotypes and genotypes are described either as case series or isolated clinical cases. This review clarifies some aspects of the phenotype and genotype of HDL, mainly HDL2, and highlights others in Africa that require further research.
Keywords
Huntington Disease Like, Africa, Phenotype, Genotype
1. Introduction
Huntington's disease is a neurodegenerative disorder similar to Huntington's disease, with which it shares the same manifestation, chorea. That latter is a hyperkinetic movement disorder characterized by an excess of brief, continuous, unpatterned involuntary movements
| [9] | BATES G, HARPER PS, JONES L (editors). Huntington’s disease, 3rd ed. Oxford: Oxford University Press; 2002. Pp. 558. S118. |
[9]
. In the last two decades, genetic plays a central role in the differential diagnosis of choreic syndromes aetiologies. HD can be the most frequent in western countries with a prevalence of up to 1 in 10,000 subjects
| [26] | WAGSTAFF LA, JOFFE R. Ataxia-telangiectasia in a South African Bantu child. S Afr Med J 1969; 43: 662-664. |
[26]
. HDL phenocopies represent about 1% of cases where HD is suspected clinically
| [3] | ANDREW SE, GOLDBERG YP, KREMER B, et al. Huntington disease without CAG expansion: phenocopies or errors in assignment? Am J Hum Genet 1994; 54: 852-863. |
| [21] | PARK JS, THORSNESS MK, POLICASTRO R et al. Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol Biol Cell 2016; 27: 2435-2449.
https://doi.org/10.1091/mbc.E16-02-0112 |
[3, 21]
. Several genetic conditions are known to cause HD phenocopies and the number of such conditions and knowledge of their clinical features has expanded in recent years. In Africa the most frequent phenocopies is HDL2 mainly described in South Africa. Other phenocopies have also been described throughout the African continent on isolated clinical cases or on series of 2 to 3 family cases. In this article we review the different clinical and genetic characteristics of HDL phenocopies in Africa to see which mutations have been described so far and how they differ clinically.
2. Methodology
Search Strategy and Search Sources
On December 2022 we conducted a systematic review on PubMed and Google Scholar. We used the terms "chorea" and the following different genes (JPH3, VPS13A, TBP, ATN1, ATXN2, ATXN7, PRNP, NBIA, SCL20A2, PDGFB, PDGFRB, XPR1, XK, C9orf72, ATM, RNF216, NKX2-1, ADCY5, PDE10A, GNAO1, FOXG1, SYT1, SCN8A), which we cross-referenced one by one with each African country.
Inclusion Criteria
- Observational epidemiological studies, case series, clinical case investigating HDL phenocopies in normal individuals and in the affected black african population and genetically confirmed
- All articles published to date
- Studies in humans
- Studies published in english and french
Exclusion Criteria
- Studies conducted in secondary choreas (vascular, infective, metabolic, ...)
- Studies conducted in HD families
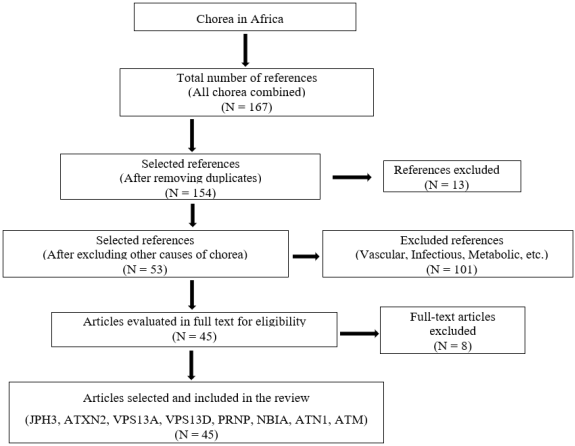
Figure 1. Flow diagram study showing case selection.
2.1. What About HD in Black Africa
HD is a severe, progressive autosomal dominant neurodegenerative disease characterized by motor, cognitive and behavioral disorders. The cause of this disease is the dynamic expansion of Cytosine-Adenine-Guanine (CAG) trinucleotides beyond their normal threshold (> 35 CAG repeats) within the Huntingtin (HTT) gene on chromosome 4 (4p16.3).
HD cases have been reported for several African countries, however, only a few of those studies are based on molecular studies and even fewer reported the allele's sizes.
Regarding this few genetic studies the reported prevalence varie from 1 to 4/100,000 inhabitants in Morocco and 0.25 to 5/100,000 inhabitants in South Africa
| [10] | BOUHOUCHE A, REGRAGUI W, LAMGHARI H, KHALDI K, BIROUK N, LYTIM S, et al. Clinical and genetic data of Huntington disease in Moroccan patients. Afri Health Sci. 2015; 15(4): 1232-1238. https://doi.org/10.4314/ahs.v15i4.23 |
| [6] | BAINE F, KRAUSE A, GREENBERG L. The frequency of Huntington disease and Huntington disease-like 2 in the South African population. Neuroepidemiology. 2016; 46(3): 198-202. https://doi.org/10.1159/000444020 |
[10, 6]
. In addition, clinical cases and case series have been reported in many other countries including two families with four patients have been reported in Burkina Faso
| [17] | KREMER B, GOLDBERG P, ANDREW SE, et al. A Worldwide Study of the Huntington’s Disease Mutation: the sensitivity and specificity of measuring CAG repeats. N Engl J Med 1994; 330: 1401-1406.
https://doi.org/10.1056/NEJM199405193302001 |
[17]
and Malian
| [14] | KABORÉ J, OUÉDRAOGO A. Huntington disease in Burkina Faso. Rev Neurol (Paris) 2000; 156(12): 1157-1158. |
[14]
, one case in Gambia
| [15] | KRAUSE A, MITCHELL C, ESSOP F, TAGER S, TEMLETT J, STEVANIN G, et al. Junctophilin 3 (JPH3) expansion mutations caus- ing Huntington disease like 2 (HDL2) are common in South African patients with African ancestry and a Huntington disease phenotype. Am J Med Genet B Neuropsychiatr Genet. 2015; 168 (7): 573-585.
https://doi.org/10.1002/ajmg.b.32332 |
[15]
and many other African countries. In the South Africa study, the largest one, the authors investigated Caucasians, mestizos and black people
| [5] | BAINE FK, KAY C, KETELAAR ME, et al. Huntington disease in the South African population occurs on diverse and ethnically distinct genetic haplotypes. Eur J Hum Genet. 2013; 21(10): 1120-1127. https://doi.org/10.1038/ejng.2014.2 |
[5]
. But the particularity in Africa is more genetic because there are more HDL than HD, the most described being HDL2. However, there are some rare cases of non-HD and non-HDL2 chorea that have been described in the continent.
2.2. How Availability Is the Neurogenetic Testing in Africa
The identification of HDL disorders requires first the exclusion of HD and then the confirmation of which HDL and then the confirmation of which HDL it is. This is not available in many African countries apart from South Africa and some North African countries like Morocco. In almost all other countries genetic studies are carried out in collaboration with institutes in Europe or the United States for molecular analysis. The study and the genetic analysis allow a positive and precise etiological diagnosis.
3. Results
From 2000 to 2022, only HD phenocopies caused by mutation of the following genes (JPH3, ATXN2, VPS13A, VPS13D, PRNP, NBIA, ATN1, ATM) were described in Africa (
Table 1). The most representative phenocopies was HDL2 (JPH3) described in case series and families in South Africa.
Table 1. Different genetic mutations, corresponding phenotypes and located country.
Genetic mutation | Phenotype and clinical features | Country |
JPH3 | choreic movements, dementia, psychiatric disorders, parkinsonian syndrome | South Africa, Botswana |
ATX2 | ataxia, choreoathetosis | South Africa |
VPS13A | choreic movements, ataxia, trunk spasm, cognitive impairment and psychiatric signs, epilepsy, dystonia, parkinsonism, dementia, seizures | Morocco, Algeria, Egypt |
PRNP | dementia, cerebral ataxia, myoclonus, choreic and choreoballic movements | Morocco, Senegal |
PKAN | ataxia, chorea, dystonia, dysmorphic syndrome, tremor, irritability, cognitive impairment, choreathetosis | Morocco, Algeria |
ATN1 | ataxia, executive dysfunction, chorea, developmental delay, epilepsy, dystonia | Sierra Leonean |
ATM | cerebellar ataxia, choreathetosis, ocular apraxia, telegectasia | Mali |
Huntington's disease-like 2/JPH3
Thirty cases of HDL2 were described, to date in black african population, of which 29 in South Africa and 1 in Botswana.
The first 3 South African cases from the same family, were described in 2007 in two males aged 53 and 42 years and in their younger sister aged 25 years. Clinically they presented, in addition to choreic movements, dementia, psychiatric disorders and a parkinsonian syndrome except for the 42 years old brother who presented myoclonus instead of parkinsonism. The age of onset was at least 4 years for the oldest subjects and up to a few months for the youngest one who had by far the highest number of repeats (59 repeats). In another series of 6 cases, published one year later still in South Africa, there were 3 male subjects with a mean age of 52.4 years with extremes ranging from 42 to 68 years and onset of the disease since 2 to 3 years. The 3 women in the series had a positive family history. Clinically, all the patients of the series presented cognitive disorders associated with choreic movements without other signs and the number of replications exceeded 42 for all of them. The largest South African series found, published in 2015, found 20 black subjects carrying the HDL2 mutation in a large cohort of whites, black and mixed-race individuals, including 3 mixed-race individuals also carrying the mutation and no mutation in the white race. In this large cohort the mean age was 51.3±9.9 (31-68) with an unknown age of onset because most patients could not give it. Symptomatically, 95% of the patients had chorea, 75% had dementia, 25% had parkinsonism and 55% had an affective disorder. The number of repercussions ranged from 45 to 58 for the whole cohort and 50% of patients had a positive family history. Elsewhere in Africa, only 1 case of JPH3, described in 2018, was found in a black subject in Botswana in a 47-year-old man, with no family history, with an evolution of the disease for 4 years. His clinical symptoms were choreic movements, slurred speech, mood instability, cognitive impairment and weight loss. He had 14/49 repeats at the mutation.
Spinocerebellar ataxie 2/ATX2
An ATX2 mutation was found, in South Africa, in a 44 years old man with choreoathetosis and bedridden for 3 months. Neither the family history nor the duration of the disease was known because of the patient's bedridden state.
Neuroacanthocytosis/VPS13A
A VPS13A mutation was described in 2 sisters of a same Moroccan family in 2020. They were 32 and 42 years old with a duration of disease evolution of 2 and 6 years respectively. They had the same mutation (homozygous c.337C > T in exon 5) but clinically they presented a difference apart from choreic movements and ataxia. In addition to these two signs the younger one presented trunk spasm, cognitive impairment and psychiatric signs while the older one presented epilepsy, dystonia and parkinsonism. The same VPS13A mutation was described in a 36-year-old Algerian man living in France with a positive family history. Clinically he had chorea, dementia, parkinsonism and seizures but the age of onset was not known. A VPS13D variant was described in a 9-year-old Egyptian girl, living in Canada, with a positive family history. She presented clinically since the age of 2 years with ataxia, cognitive impairment, chorea.
Prion protein/PRNP
Two cases of PRNP mutation were described in Morocco in a 54-year-old man living in France and in Senegal in a 62-year-old man respectively in 1999 and in 2022 with for the Moroccan one the V210I mutation and the presence of the 14-3-3 protein in the CSF for both cases. Clinically, both patients presented dementia, cerebral ataxia and myoclonus in addition to choreic movements for the Moroccan patient and choreoballic movements for the Senegalese patient. Both patients had no family history.
NBIA/PKAN (Pantothenate kinase-associated neurodegen eration)
In 2017, NBIA was described in Algeria in two brothers of the same family aged 21 and 18 years. The onset of their disease was 5 years for the older brother and 8 years for the younger brother. Clinically they both had ataxia, chorea and dystonia with in addition in the older brother dysmorphic syndrom and in the younger brother a tremor, irritability. Another case of NBIA was described in 2020 in a 10 years old Moroccan girl living in the UK. She had no family history and presented since the age of 16 months in a slowly progressive way a cognitive impairment, ataxia, dystonia, choreathetosis.
Atrophin-1/ATN1
In a Sierra Leonean family living in the USA, a 50-year-old man and his 11-year-old niece and 7-year-old nephew were described in 2019 with the same ATN1 mutation (61 CAG in allele 1). They had a positive family history and the man presented ataxia, executive dysfunction, chorea while the children presented developmental delay and epilepsy in addition to dystonia in the nephew.
AT3/ATM
In Mali in 2013, three individuals in the same family (homozygous T7985A). That family was composed of male subjects aged 14, 10, and 2 years with an age of onset of at most 2 years for all of them, of which the oldest presented cerebellar ataxia, choreathetosis, ocular apraxia, telegectasia, and the youngest presented only cerebellar ataxia.
4. Discussion
On this review we have narrated the main HDL syndromes described nowadays in the black African subject.
While HD is clearly documented to be present in many parts of the European and US countries, some HD-like diseases as HDL2 are more frequent in african people.
It is clear from this review that there is a clear difference between northern and southern africa, especially sub-Saharan Africa, in terms of the genotypic and phenotypic distribution of chorea. The northern part is more represented due to the lack of adequate human and technical resources in the field of movement disorders in the southern part outside South Africa.
In black South African patients who tested positive for an HD-causing mutation, approximately 35% had a JPH3 mutation and 65% an HD mutation
| [8] | BARDIEN S, ABRAHAMS F, SOODYALL H, MERWE LVD, GREENBERG J, BRINK T, CARR J. A South African Mixed Ancestry Family with Huntington Disease-Like 2: Clinical and Genetic Features. Movement Disorders 2007; 22(14): 2083-2089. https://doi.org/10.1002/mds.21672 |
[8]
. This added further weight to the hypothesis that HDL2 is an African disease. Larger studies have confirmed the high frequency of JPH3 mutations in patients of African ancestry with a HD phenotype
| [1] | ABASCAL JV, ALVAREZ PB, ROSALES FM, RAMIREZ AF, KHALIL MIA, VOLPINI LJLV, NYAN O. First case with Huntington's disease in The Gambia. Journal of the Neurological Sciences 2014; 338: 238-240.
https://doi.org/10.1016/j.jns.2013.12.045 |
[1]
. In our review we found 30 cases of HDL2 described in the southern regions of Africa. There were no ethnic groups or tribes with a particularly high prevalence of HDL2, although this deserves investigation. But we believe that the lack of human resources and adequate technical facilities may contribute to this gap between South Africa and other parts of the continent. This disparity is even seen in South Africa where 80% of the population is black and the prevalence of HD is higher among whites, suggesting limited access to care for the black population. It is likely that this is a result of poor access to specialist medical services; however, underlying genetic variation may be a key factor
. Concerning the number of Cytosine-Thymine-Guanine (CTG) repeats we noted that there is a positive correlation inversely proportional with age
| [7] | BAINE FK, PEERBHAI N, KRAUSE A. A study of Huntington disease-like syndromes in black South African patients reveals a single SCA2 mutation and a unique distribution of normal alleles across five repeat loci. Journal of the Neurological Sciences 2018; 390: 200-204.
https://doi.org/10.1016/j.jns.2018.04.031 |
[7]
. Younger subjects have a higher number of repeats and certainly a faster progression of the disease.
Until now, only spinocerebellar ataxia (SCA) 17 was known to mimic HD. Although HD rarely presents as pure cerebellar ataxia, it is a common feature of the disease
| [13] | GREGORY A, POLSTER BJ, HAYFLICK SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet 2008; 46(2): 73-80.
https://doi.org/10.1136/jmg.2008.061929 |
[13]
. Many of the autosomal dominant spinocerebellar ataxias (SCAs) produce extracerebellar features and have phenotypic overlap with Huntington’s disease. SCA17 is the closest clinical mimic of Huntington’s disease and the commonest identified cause of Hunting- ton’s disease phenocopy syndromes
| [11] | COULIBALY T, KARAMBE M, GUINTO CO, TRAORE M. Huntington's disease confirmed by genetic testing in three Malian families. Mov Disord 2012; 27(Suppl.1): 171. |
[11]
. We found on this study the only case of SCA2
| [4] | ASSAMI S, AZZEDINE H, NOUIOUA S, MUNDWILLER E, MAHOUI S, MAKRI S, DJEMAI M, GRID D, BRICE A, HAMADOUCHE T, STEVANIN G, TAZIR M. Pantothenate Kinase–Associated Neurodegeneration: Clinical Description of 10 Patients and Identification of New Mutations. Movement Disorders 2011: 26 (9): 1777-1779.
https://doi.org/10.1002/mds.23648 |
[4]
, with an HDL phenotype as choreathetoisis, described so far has no particularity on the repeats (roughly equal to the other SCA2 described). Moreover, it was a young patient who was bedridden for 3 months. Did she have other co-morbidities? Or did she have a more rapid progression of the disease? This deserves more attention to find an explanation certainly by making a comparison with SCA2 without choreic phenotype.
Neuroacanthocytosis, an autosomal recessive disorder, has been very rarely reported in Africa and never in black people to our knowledge. Only 2 cases from the same family have been reported in Morocco and 2 other cases of maghrebian origin have been described in the West. The particularity lies mainly in the clinical phenotypes which are quite varied with epileptic seizures and parkinsonism which are not constant. This phenotypic variability has been already documented in many cases of Choreacanthocytosis (ChAc) even in twins
| [19] | LANDOURE G, MOCHEL F, MEILLEUR K, LY M, SANGARE M, BOCOUM N, BAGAYOKO K, COULIBALY T, SARR AM, BA HO, COULIBALY S, GUINTO CO, TOURE M, TRAORE M, FISCHBECK KH. Novel mutation in the ATM gene in a Malian family with ataxia telangiectasia. J Neurol 2013; 260: 324-326. |
[19]
and suggests the implication of other modifier genes, epigenetic and environmental factors on the pathogenesis of neuroacanthocytosis as discussed elsewhere. The underlying pathophysiology of ChAc is not yet well known, but recently it was shown that chorein may have a role in lipid exchange between organelles and thus for membrane lipid homeostasis in the nervous system. n deed, it has shown that N-terminal region of Vps13A, called Chorein_N domain, is a lipid transport module that bind to the endoplasmic reticulum and connects it to mitochondria
| [23] | STEVANIN G, FUJIGASAKI H, LEBRE A-S, et al. Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 2003; 126: 1599-1603. https://doi.org/10.1093/brain/awg155 |
[23]
. Unlike VPS13A, the VPS13D variant described in children is more characterized by developmental disorders. In contrast to VPS13A, the VPS13D variant described in children is more characterized by a global developmental delay that are absent, at least at young ages, in variant A. The VPS13D clinical spectrum suggests mitochondrial dysfunction as a possible pathophysiological mechanism for this new clinical entity
| [12] | DONALDSON I, MARSDEN CD, SCHNEIDER SA, BHATIA KB. Clinical approach to movement disorders. In Marsden's Book of Movement Disorders. Donaldson I, Marsden CD, Schneider SA, Bhatia KB. Editors. Oxford, United Kingdom: Oxford University Press; 2012: 140-141. |
[12]
. The VPS13D clinical spectrum includes corticospinal tract dysfunction, cerebellar and extrapyramidal signs, hearing loss, and seizures together with bilateral symmetric T2 hyperintensities in the basal ganglia and/or brainstem. Mitochondrial leukodystrophies also display a pattern of diffuse asymmetrical subcortical white matter and bilateral basal ganglia involvement
| [12] | DONALDSON I, MARSDEN CD, SCHNEIDER SA, BHATIA KB. Clinical approach to movement disorders. In Marsden's Book of Movement Disorders. Donaldson I, Marsden CD, Schneider SA, Bhatia KB. Editors. Oxford, United Kingdom: Oxford University Press; 2012: 140-141. |
[12]
.
The PRNP mutation has never been found in Africa. Of the two cases found in this review, one was described in a Moroccan man living in France and the other in a Senegalese man in whom the diagnosis was made on the basis of clinical symptoms and the presence of the 14-3-3 protein in the cerebrospinal fluid. This rarity in Africa does not necessarily mean an absence of the disease but certainly an under-diagnosis due to the limited access to care; in many African countries, the countries or their families pay for the care themselves, as was the case for the Senegalese patient. Clinically the two cases are similar with ataxia, dementia, myoclonus and choreic or choreoballic movements.
Usually described in young subjects, PKAN may have a slightly later age of onset (around the second decade). Especially described in the Maghreb countries, especially in Algeria where 10 other cases of PKAN have been described in 3 families but without choreic movements
| [20] | MAGAZI DS, KRAUSE A, BONEV V, MOAGI M, IQBAL Z, DLUDLA M, VAN DER MEYDEN CH. Huntington's disease: genetic heterogeneity in black African patients. S Afr Med J 2008; 98: 200-203. |
[20]
. PKAN is characterized by dystonia, parkinsonism and iron accumulation in the brain, and accounts for around half of cases of neurodegeneration with brain iron accumulation (NBIA), a group of progressive neurodegenerative disorders characterized by high levels of iron, and the presence of axonal spheroids, usually limited to the brain and central nervous system
| [2] | AIYESIMOJU AB, OSUNTOKUN BO, BADEMOSI O, ADEUJA AO. Hereditary neurodegenerative disorders in Nigerian Africans. Neurology 1984; 34: 361-362.
https://doi.org/10.1212/wnl.34.3.361 |
[2]
. Forms with choreic movements are very rarely reported. The two Algerian cases that we found with choreic movements were not genetically typed (due to the non-availability of a specialized laboratory) but the other large Algerian series of 10 cases (in collaboration with a Parisian genetic laboratory) showed a hyperactivity present in all patients and frequent falls in half of the patients and the homozygous truncating PANK2 mutations found was c.846_847delAG/p.S282SfsX3 in exon 3 and c.1171_1174dupATTG/p.G392DfsX11 in exon 5
| [20] | MAGAZI DS, KRAUSE A, BONEV V, MOAGI M, IQBAL Z, DLUDLA M, VAN DER MEYDEN CH. Huntington's disease: genetic heterogeneity in black African patients. S Afr Med J 2008; 98: 200-203. |
[20]
.
Atrophin-1 is not at all common in Africa, the 3 cases described were in a Sierra Leonean family living in the USA.
On the other hand, with a north-south collaboration, Malian authors described cases of ATM (homozygous T7985A mutation) in 3 persons of the same family with a choreoathetosis movement phenotype
| [16] | KRAUSE A, TEMLETT J, VAN DER MEYDEN K, ROSS CA, CALLAHAN C, MARGOLIS RL. CAG/CTG repeat expansions at the HDL2 locus are a common cause of Huntington disease in black South Africans. Am J Hum Genet. 2002; 71(4): 528-528. |
[16]
. Although cases of A-T have been reported in populations in North Africa
| [24] | TRIKI C, FEKI I, MEZIOU M, TURKI H, ZAHAF A, MHIRI C. Clinical, biological and genetic study of 24 patients with ataxia-telangiectasia from southern Tunisia. Rev Neurol 2000; 156: 634-637. |
[24]
but reports of this disease in sub-Saharan Africa have been limited to clinical characterization
| [18] | KUMAR N, LEONZINO M, HANCOCK-CERUTTI W, HORENKAMP FA, LI P, LEES JA et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol. 2018; 217(10): 3625-3639.
https://doi.org/10.1083/jcb.201807019 |
| [25] | VELAYOS BAEZA A, DOBSON-STONE C, RAMPOLDI L, BADER B, WALKER RH, DANEK A, et al. Chorea-Acanthocytosis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, Amemiya A, editors. Gene Reviews. Seattle: Seattle (WA): University of Washington; 2002. p. 1993-2019. [updated 2019 Apr 18]. |
[18, 25]
.
There are a limited number of studies on HDL in Africa. Most of the studies have been done in South Africa or in the Maghreb and the few other cases described in Sub-Saharan Africa are done through collaboration with Western laboratories. Moreover, the few studies carried out in the continent often show exclusively African genotypes and phenotypes such as HDL2. Pure choreic phenotypes are more prevalent in the Maghreb and South Africa, while mixed phenotypes, especially those with predominant ataxia, are more common in certain southern countries. Large-scale studies in most African countries could confirm this assertion.
5. Conclusion
Limited number of HD-like disorders have been identified in black african population. Many patients are likely undiagnosed for a number of reasons, including lack of access to care and lack of specialists in the field. Geographically, phenotypes and genotypes are distributed unevenly across Africa, with a clear predominance in South Africa and the Maghreb, where pure choreic forms are more common. However, Africa is full of interesting phenotypes that deserve to be better investigated especially with the demographic transition that this continent is undergoing.
Abbreviations
JPH3 | JunctoJphilin-3 |
ATXN2 | Ataxine 2 |
VPS13A | Vacuolar Protein Sorting 13 Homolog A |
VPS13D | Vacuolar Protein Sorting 13 Homolog D |
PRNP | Prion Protein |
NBIA | Neurodegeneration with Brain Iron Accumulation |
ATN1 | Atrophin 1 |
ATM | Ataxia-telangiectasia Mutated |
Author Contributions
Fall Maouly: Conceptualization, Data curation, Investigation Methodology, Visualization, Writing – original draft, Writing – review & editing
Diop Alassane Mamadou: Visualization, Writing – review & editing
Kahwagi Jamil: Visualization, Writing – review & editing
Gaye Ndiaga Matar: Visualization, Writing – review & editing
Mie Rizig: Visualization, Writing – review & editing
Conflicts of Interest
The authors declare no conflicts of interest.
References
| [1] |
ABASCAL JV, ALVAREZ PB, ROSALES FM, RAMIREZ AF, KHALIL MIA, VOLPINI LJLV, NYAN O. First case with Huntington's disease in The Gambia. Journal of the Neurological Sciences 2014; 338: 238-240.
https://doi.org/10.1016/j.jns.2013.12.045
|
| [2] |
AIYESIMOJU AB, OSUNTOKUN BO, BADEMOSI O, ADEUJA AO. Hereditary neurodegenerative disorders in Nigerian Africans. Neurology 1984; 34: 361-362.
https://doi.org/10.1212/wnl.34.3.361
|
| [3] |
ANDREW SE, GOLDBERG YP, KREMER B, et al. Huntington disease without CAG expansion: phenocopies or errors in assignment? Am J Hum Genet 1994; 54: 852-863.
|
| [4] |
ASSAMI S, AZZEDINE H, NOUIOUA S, MUNDWILLER E, MAHOUI S, MAKRI S, DJEMAI M, GRID D, BRICE A, HAMADOUCHE T, STEVANIN G, TAZIR M. Pantothenate Kinase–Associated Neurodegeneration: Clinical Description of 10 Patients and Identification of New Mutations. Movement Disorders 2011: 26 (9): 1777-1779.
https://doi.org/10.1002/mds.23648
|
| [5] |
BAINE FK, KAY C, KETELAAR ME, et al. Huntington disease in the South African population occurs on diverse and ethnically distinct genetic haplotypes. Eur J Hum Genet. 2013; 21(10): 1120-1127.
https://doi.org/10.1038/ejng.2014.2
|
| [6] |
BAINE F, KRAUSE A, GREENBERG L. The frequency of Huntington disease and Huntington disease-like 2 in the South African population. Neuroepidemiology. 2016; 46(3): 198-202.
https://doi.org/10.1159/000444020
|
| [7] |
BAINE FK, PEERBHAI N, KRAUSE A. A study of Huntington disease-like syndromes in black South African patients reveals a single SCA2 mutation and a unique distribution of normal alleles across five repeat loci. Journal of the Neurological Sciences 2018; 390: 200-204.
https://doi.org/10.1016/j.jns.2018.04.031
|
| [8] |
BARDIEN S, ABRAHAMS F, SOODYALL H, MERWE LVD, GREENBERG J, BRINK T, CARR J. A South African Mixed Ancestry Family with Huntington Disease-Like 2: Clinical and Genetic Features. Movement Disorders 2007; 22(14): 2083-2089.
https://doi.org/10.1002/mds.21672
|
| [9] |
BATES G, HARPER PS, JONES L (editors). Huntington’s disease, 3rd ed. Oxford: Oxford University Press; 2002. Pp. 558. S118.
|
| [10] |
BOUHOUCHE A, REGRAGUI W, LAMGHARI H, KHALDI K, BIROUK N, LYTIM S, et al. Clinical and genetic data of Huntington disease in Moroccan patients. Afri Health Sci. 2015; 15(4): 1232-1238.
https://doi.org/10.4314/ahs.v15i4.23
|
| [11] |
COULIBALY T, KARAMBE M, GUINTO CO, TRAORE M. Huntington's disease confirmed by genetic testing in three Malian families. Mov Disord 2012; 27(Suppl.1): 171.
|
| [12] |
DONALDSON I, MARSDEN CD, SCHNEIDER SA, BHATIA KB. Clinical approach to movement disorders. In Marsden's Book of Movement Disorders. Donaldson I, Marsden CD, Schneider SA, Bhatia KB. Editors. Oxford, United Kingdom: Oxford University Press; 2012: 140-141.
|
| [13] |
GREGORY A, POLSTER BJ, HAYFLICK SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet 2008; 46(2): 73-80.
https://doi.org/10.1136/jmg.2008.061929
|
| [14] |
KABORÉ J, OUÉDRAOGO A. Huntington disease in Burkina Faso. Rev Neurol (Paris) 2000; 156(12): 1157-1158.
|
| [15] |
KRAUSE A, MITCHELL C, ESSOP F, TAGER S, TEMLETT J, STEVANIN G, et al. Junctophilin 3 (JPH3) expansion mutations caus- ing Huntington disease like 2 (HDL2) are common in South African patients with African ancestry and a Huntington disease phenotype. Am J Med Genet B Neuropsychiatr Genet. 2015; 168 (7): 573-585.
https://doi.org/10.1002/ajmg.b.32332
|
| [16] |
KRAUSE A, TEMLETT J, VAN DER MEYDEN K, ROSS CA, CALLAHAN C, MARGOLIS RL. CAG/CTG repeat expansions at the HDL2 locus are a common cause of Huntington disease in black South Africans. Am J Hum Genet. 2002; 71(4): 528-528.
|
| [17] |
KREMER B, GOLDBERG P, ANDREW SE, et al. A Worldwide Study of the Huntington’s Disease Mutation: the sensitivity and specificity of measuring CAG repeats. N Engl J Med 1994; 330: 1401-1406.
https://doi.org/10.1056/NEJM199405193302001
|
| [18] |
KUMAR N, LEONZINO M, HANCOCK-CERUTTI W, HORENKAMP FA, LI P, LEES JA et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol. 2018; 217(10): 3625-3639.
https://doi.org/10.1083/jcb.201807019
|
| [19] |
LANDOURE G, MOCHEL F, MEILLEUR K, LY M, SANGARE M, BOCOUM N, BAGAYOKO K, COULIBALY T, SARR AM, BA HO, COULIBALY S, GUINTO CO, TOURE M, TRAORE M, FISCHBECK KH. Novel mutation in the ATM gene in a Malian family with ataxia telangiectasia. J Neurol 2013; 260: 324-326.
|
| [20] |
MAGAZI DS, KRAUSE A, BONEV V, MOAGI M, IQBAL Z, DLUDLA M, VAN DER MEYDEN CH. Huntington's disease: genetic heterogeneity in black African patients. S Afr Med J 2008; 98: 200-203.
|
| [21] |
PARK JS, THORSNESS MK, POLICASTRO R et al. Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol Biol Cell 2016; 27: 2435-2449.
https://doi.org/10.1091/mbc.E16-02-0112
|
| [22] |
ROSS CA, TABRIZI SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011; 10: 83-98.
https://doi.org/10.1016/S1474-4422(10)70245-3
|
| [23] |
STEVANIN G, FUJIGASAKI H, LEBRE A-S, et al. Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 2003; 126: 1599-1603.
https://doi.org/10.1093/brain/awg155
|
| [24] |
TRIKI C, FEKI I, MEZIOU M, TURKI H, ZAHAF A, MHIRI C. Clinical, biological and genetic study of 24 patients with ataxia-telangiectasia from southern Tunisia. Rev Neurol 2000; 156: 634-637.
|
| [25] |
VELAYOS BAEZA A, DOBSON-STONE C, RAMPOLDI L, BADER B, WALKER RH, DANEK A, et al. Chorea-Acanthocytosis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, Amemiya A, editors. Gene Reviews. Seattle: Seattle (WA): University of Washington; 2002. p. 1993-2019. [updated 2019 Apr 18].
|
| [26] |
WAGSTAFF LA, JOFFE R. Ataxia-telangiectasia in a South African Bantu child. S Afr Med J 1969; 43: 662-664.
|
Cite This Article
-
APA Style
Maouly, F., Mamadou, D. A., Jamil, K., Matar, G. N., Moustapha, N., et al. (2025). Huntington Disease Like in Black African Population:
A Clinical and Genetical Review. Clinical Neurology and Neuroscience, 9(4), 73-79. https://doi.org/10.11648/j.cnn.20250904.14
 Copy
|
Copy
|
 Download
Download
ACS Style
Maouly, F.; Mamadou, D. A.; Jamil, K.; Matar, G. N.; Moustapha, N., et al. Huntington Disease Like in Black African Population:
A Clinical and Genetical Review. Clin. Neurol. Neurosci. 2025, 9(4), 73-79. doi: 10.11648/j.cnn.20250904.14
Copy
|
Download
AMA Style
Maouly F, Mamadou DA, Jamil K, Matar GN, Moustapha N, et al. Huntington Disease Like in Black African Population:
A Clinical and Genetical Review. Clin Neurol Neurosci. 2025;9(4):73-79. doi: 10.11648/j.cnn.20250904.14
Copy
|
Download
-
@article{10.11648/j.cnn.20250904.14,
author = {Fall Maouly and Diop Alassane Mamadou and Kahwagi Jamil and Gaye Ndiaga Matar and Ndiaye Moustapha and Mie Rizig},
title = {Huntington Disease Like in Black African Population:
A Clinical and Genetical Review},
journal = {Clinical Neurology and Neuroscience},
volume = {9},
number = {4},
pages = {73-79},
doi = {10.11648/j.cnn.20250904.14},
url = {https://doi.org/10.11648/j.cnn.20250904.14},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.cnn.20250904.14},
abstract = {Huntington Disease-like (HDL) is a neurodegenerative disorder similar to Huntington Disease (HD) in its clinical phenotype, genetic characteristics, neuropathology and longitudinal progression. We review the different phenocopies of HDL in Africa from a clinical and genetic perspective through published cases. A literature review through PubMed and Google Scholar of all clinically and genically described cases of HDL until the end of December 2022 was performed and a descriptive analysis was carried out. Fifteen papers were published from 2000 to 2022 in Africa on HDL. Only HD phenocopies caused by mutation of the following genes (JPH3, ATXN2, VPS13A, VPS13D, PRNP, NBIA, ATN1, ATM) were described. The most representative phenocopies was HDL2 (JPH3) described in case series and families in South Africa. Other phenotypes and genotypes are described either as case series or isolated clinical cases. This review clarifies some aspects of the phenotype and genotype of HDL, mainly HDL2, and highlights others in Africa that require further research.},
year = {2025}
}
Copy
|
Download
-
TY - JOUR
T1 - Huntington Disease Like in Black African Population:
A Clinical and Genetical Review
AU - Fall Maouly
AU - Diop Alassane Mamadou
AU - Kahwagi Jamil
AU - Gaye Ndiaga Matar
AU - Ndiaye Moustapha
AU - Mie Rizig
Y1 - 2025/12/29
PY - 2025
N1 - https://doi.org/10.11648/j.cnn.20250904.14
DO - 10.11648/j.cnn.20250904.14
T2 - Clinical Neurology and Neuroscience
JF - Clinical Neurology and Neuroscience
JO - Clinical Neurology and Neuroscience
SP - 73
EP - 79
PB - Science Publishing Group
SN - 2578-8930
UR - https://doi.org/10.11648/j.cnn.20250904.14
AB - Huntington Disease-like (HDL) is a neurodegenerative disorder similar to Huntington Disease (HD) in its clinical phenotype, genetic characteristics, neuropathology and longitudinal progression. We review the different phenocopies of HDL in Africa from a clinical and genetic perspective through published cases. A literature review through PubMed and Google Scholar of all clinically and genically described cases of HDL until the end of December 2022 was performed and a descriptive analysis was carried out. Fifteen papers were published from 2000 to 2022 in Africa on HDL. Only HD phenocopies caused by mutation of the following genes (JPH3, ATXN2, VPS13A, VPS13D, PRNP, NBIA, ATN1, ATM) were described. The most representative phenocopies was HDL2 (JPH3) described in case series and families in South Africa. Other phenotypes and genotypes are described either as case series or isolated clinical cases. This review clarifies some aspects of the phenotype and genotype of HDL, mainly HDL2, and highlights others in Africa that require further research.
VL - 9
IS - 4
ER -
Copy
|
Download